Step 8: Collect your data for regulatory submission
Healthcare product development is complex. It is critical to know the general product development lifecycle so that you can follow the correct path. Knowing which pitfalls to avoid will speed your time to market. This article outlines step 8 of the 10 steps to develop a healthcare product.
When developing a healthcare product, you will collect scientific data from various sources, including chemistry, manufacturing and control (CMC) and design activities, as well as necessary biopharmaceutical, non-clinical and clinical studies. All these data should be organized, summarized and analyzed so that the licence or clearance application is complete, transparent and unambiguous and will facilitate regulatory review and approval or clearance of the healthcare product. Although the data will be obtained only after the execution of each study, it is at the planning stage for each study that the data presentation should be defined (that is, what to collect, and how to present it). Note that planning ahead will help you achieve the results you want.1
Healthcare product development: What data to collect?
The type of data and information collected will be summarized in your product label. To ensure the completeness of the data being collected1,2, plan for the content of the study reports and submission summaries* while you are at the stage of the study design or protocol planning. A careful review of applicable guidelines that discuss the format and content of submission documents is thus extremely important. This information can be gathered by reviewing documents published by the International Conference on Harmonisation (ICH), different regulatory agencies (such as Health Canada, the U.S. Food and Drug Administration [FDA] and the European Medicines Agency [EMA]), as well as other recognized standards.
Examples of these documents may include clinical study reports, clinical protocols/plans, Quality Overall Summaries, Non-Clinical Summaries, Clinical Summaries, and integrated summaries of safety and effectiveness. As well, many product-specific guidance documents (for example, for anti-cancer medicinal products, antiviral drugs) are available that address different types or phases of product development.
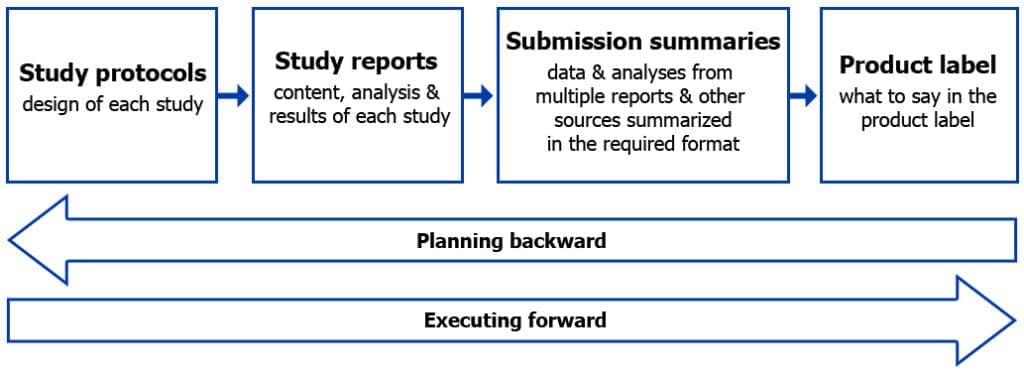
Figure 1 illustrates the relationship between the different regulatory documents, including study protocols, study reports, submission summaries and the product label.
Figure 1: Product development: Planning backward while executing forward
Using templates
Information in a regulatory submission should be presented in a logical and consistent manner that will facilitate regulatory review. A high-quality and professional submission helps assure regulatory reviewers that the submission has been carefully prepared and that any concerns have been appropriately addressed.
One way to achieve a well-organized submission is through the use of templates. These templates should be created and they should:
- Be user-friendly (for example, with suggested wordings or examples of how to present tables and figures)
- Provide references to the applicable guidance documents so that the author(s) can understand the basis for the data requirements and presentation format
- Include a built-in style for how each document should be presented (for example, headings, headers, footers, pagination, table of contents)
The use of templates is particularly useful to help generate summaries (such as an integrated summary of safety) using data from multiple study reports.
Conclusion
The process of establishing the specific content and format of each regulatory document included in a clearance or licensing application can be lengthy and difficult. Nevertheless, this process allows the developer to ensure the completeness of the data being collected and it facilitates the preparation of the regulatory submission. “Doing it right” at the beginning also helps expedite the regulatory review and approval process.
*Examples of regulatory summaries include the executive summary for medical devices, and the Quality Overall Summary, Non-Clinical Summary and Clinical Summary for drugs or biologics.
Read the next step in the 10 steps to develop a healthcare product: Step 9: Collate your regulatory submission
Read the previous step: Step 7: Execute your clinical plan
Disclaimer
The information presented in these articles is intended to outline the general processes, principles and concepts of the healthcare product development lifecycle. Since regulatory requirements are ever-changing, it is current only as of the date of publication and not intended to provide detailed instructions for product development. Every healthcare product is unique and therefore so is its associated product development lifecycle. Specific advice should be sought from a qualified healthcare or other appropriate professional.
Published: October 17, 2012
References
- McPhatter, K., Walch, K., Miller L. & Kneifel, A. (1999). Chapter 27. Assembling and filing the NDA/BLA. In S.E. Linberg (Ed.) Expediting drug and biologics development: A strategic approach. (n.d.) Watham, MA: PAREXEL International.
- Granzer, U. (2001). Getting the foundation right. Prescribing information, summary of product characteristics, package leaflet. British Institution for Regulatory Affairs (BIRA) Module 6. Regulatory Strategy: The Market Place.