Step 2: Identify your healthcare claim and product label
Healthcare product development is complex. It is critical to know the general product development lifecycle so that you can follow the correct path. Knowing which pitfalls to avoid will speed your time to market. This article outlines step 2 of the 10 steps to develop a healthcare product.
A claim or an indication of a healthcare product is a simple and concise statement of the condition(s) under which a drug, biologic or medical device will be used. It normally has the following format:
[Product X] is to diagnosis, cure, mitigate, treat or prevent a certain disease or condition.
Although a simple statement, it is, in fact, the essence of all the facts and scientific data collected throughout the many years spent in the product development.1
Your data and the healthcare product label
How are the collected data used to support this statement?
For most healthcare products,i the scientific data are compiled in a specific format and submitted in a licence applicationii for regulatory clearance or approval. The regulatory body then reviews the application and may raise questions for the applicant to address or clarify.
The product label that is cleared or approved typically includes the company details and product information (a product description) as well as information on the uses for which the healthcare product has been shown to be effective, any associated risks and how to use the product. For example, labelling for a prescription drug in the US represents a summary of all the non-clinical and clinical studies conducted over the period of years from drug discovery through product development to approval by the U.S. Food and Drug Administration (FDA). The essential prescribing information for a prescription drug for use in humans is provided in the package insert, which states the usefulness and the risks associated with the product to ensure safe and effective use. All marketing activities must be consistent with the claim and the information presented on this label.1,2
At the end of the review, if the agency concludes that the data supplied in the application substantiates the proposed claim from the standpoints of safety, efficacy (or effectiveness) and quality, then an approval or clearance is issued. A regulatory approval or clearance is for the combination of the product and its label (including product claim).
Keep the end product in mind
To have a successful and effective healthcare product development program, it is critical for a developer to identify the desired end product (that is, what the intended claim and product label will say) right from the beginning. This way, all information that will need to be collected during product development can be identified and properly procured.
To illustrate this point, the figure below shows how the information collected during product development is organized and presented in a licence application.
Figure 1: From raw data to product label (including product claim)
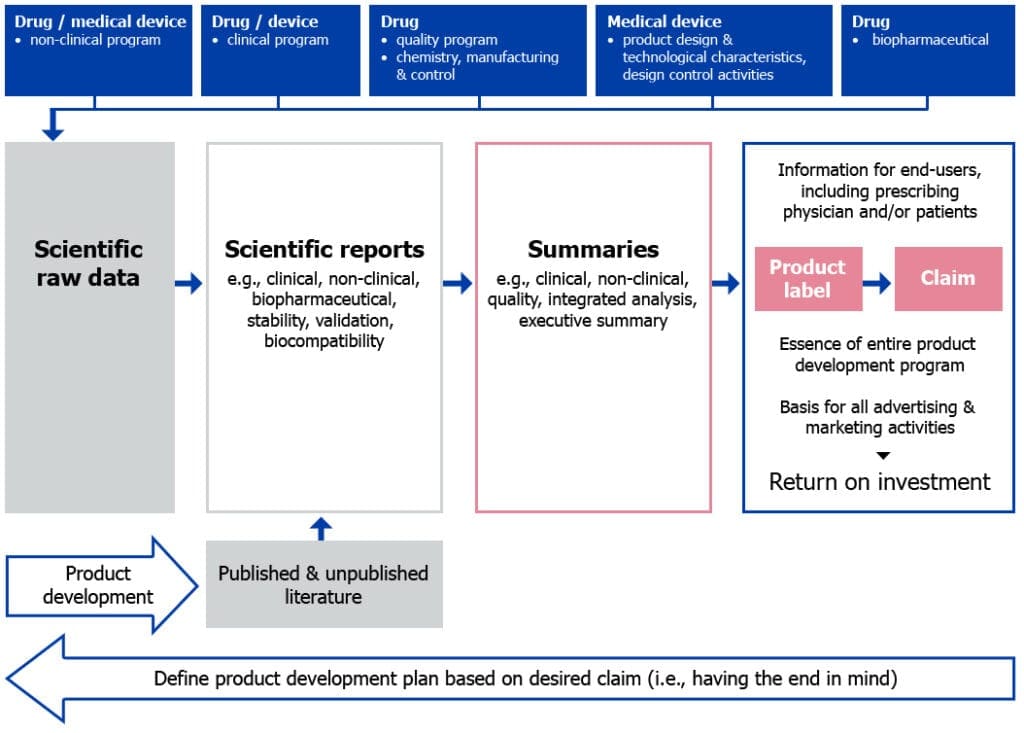
- During the product development phase, scientific raw data are collected from clinical, non-clinical, biopharmaceutical and manufacturing or design control activities.
- Raw data are then organized, analyzed and presented in various scientific reports, which may also be supported by any published and/or unpublished literature.
- Scientific reports are further summarized into different types of clinical, non-clinical and quality summaries, or integrated analysis or executive summaries, as required by different types of licence applications.
- Finally, summaries are abridged to the essential information on the drug, biologic or medical device and presented as the product label, which also includes the claim statement.
Since the product label is ultimately the document that is presented to the end-users and is the basis for advertising and marketing activities (that is, the return on investment), careful attention and investment should be paid to it in order to allow it to drive the product development program.
Did you know?
Sometimes, the product claim is vital in determining how some healthcare products are regulated, which in turn dictates the level of scrutiny to which they may be subjected. For example, in the US, a device such as software that analyzes MRIiii images is designated as a class II 510(k) product if it only measures the size or volume of anatomical structures. However, if the software detects abnormalities or provides diagnostic information, it would be considered a class III PMA (premarket approval) device. A medical device developer may choose to “start small” and begin interacting with the FDA with a simpler 510(k) before moving to a more challenging PMA once a revenue stream is established.3
iIn the US, some products (for example, low-risk class I devices or 510(k) exempt devices, regulatory review is not required prior to the commercialization of the product.
iiLicence applications can be made for drugs, biologics or medical devices, such as a Canadian New Drug Submission (NDS), a US New Drug Application (NDA), an EU Marketing Authorization Application (MAA), a US Biological License Application (BLA), a US 510(k) application, a US premarket approval (PMA) or a Canadian medical device licence application.
iiiMRI = magnetic resonance imaging
Read the next step in the 10 steps to develop a healthcare product: Step 3: Determine your healthcare market
Read the previous step: Step 1: Classify your healthcare product
Disclaimer
The information presented in these articles is intended to outline the general processes, principles and concepts of the healthcare product development lifecycle. Since regulatory requirements are ever-changing, it is current only as of the date of publication and not intended to provide detailed instructions for product development. Every healthcare product is unique and therefore so is its associated product development lifecycle. Specific advice should be sought from a qualified healthcare or other appropriate professional.
Published: October 17, 2012
References
- Ansel, H.C., Popovich, N.C. & Allen, L.V. Jr. (1995). Chapter 2. New drug development and approval process. In Pharmaceutical dosage forms and drug delivery systems (6th ed.). Baltimore: A Waverly Company.
- Tobin, J.J. & Walsh, G. (2008). Chapter 12. Oversight and Vigilance. In Medical product regulatory affairs. Pharmaceuticals, diagnostics, medical devices. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA.
- Pisano, D.J. & Mantus, D.S. (Eds.). (2008). FDA Regulatory Affairs: A Guide for Prescription Drugs, Medical Devices, and Biologics (2nd ed.). New York, NY: Informa Healthcare.