Medical device submissions: Procedures to legally place a medical device on the market
The procedures required to legally place a medical device on the market vary in Canada, the US and the EU. Different regulatory bodies are involved, requiring different types of submissions (Table 1) and review timelines depending on the classification of the device.
Table 1: Medical device submissions: Canada, the US and the EU1–9
| Jurisdiction | Submission | Device classification | Regulatory body |
| Canada | Establishment registration (MDEL) | Class I & all importers or distributors of all four device classes to permit importation or distribution (sale) of a medical device in Canada1 | Health Canada |
| Medical Device Licence Application | Classes II, III & IV | ||
| US | Establishment registration | Classes I, II & III | U.S. Food and Drug Administration (FDA) |
| Traditional, abbreviated or special 510(k)s | Class I, II, and III (where PMA is not required) | ● FDA
OR ● Accredited party + FDA (for products eligible for third-party review) |
|
| De novo | Low- to moderate-risk devices that have been classified as class III because they were found not substantially equivalent to existing devices | FDA | |
| PMA/PDP* | Class III & high-risk devices that are non-substantially equivalent (NSE) to class I, II or III (device may be eligible for de novo)2 | FDA | |
| EU | Technical documentation | Class I
Exceptions: Class Is*, Im*, Ir* |
Self-certification by manufacturer |
| Technical file | Class Im, Is, Ir, IIa, IIb, III | Notified Body | |
| Design dossier | Class III | Notified Body | |
| Consultation dossier (only for the medicinal ingredient used in the device) | Class III (a device with a medicinal ingredient)
and Class IIa and IIb if they compose of substances or a combination of substances to be introduced to the body
|
Competent Authority or European Medicines Agency (EMA) coordinated via the Notified Body |
*PMA = Premarket authorization application
PDP = Product development protocol
Class Is = Class I sterile
Class Im = Class I measuring
Class Ir = Class I reusable surgical instruments
Canada: Registration process for medical devices
The regulatory review process in Canada is straightforward. The sponsor (applicant) deals with a single regulatory agency, Health Canada. For class I medical devices, the manufacturer needs to apply for a Medical Device Establishment Licence (MDEL) unless he or she imports or distributes it solely through someone who already holds an MDEL. For medical devices that are classified as class II, III or IV, a Medical Device Licence application must be submitted.
The higher the risk of the medical device, the more information is required in the licence application and the more the time is required to reach regulatory approval (for example, in the first screening and review cycle, the agency’s performance goals are 15 days for class II and 90 days for class IV).
The submission typically goes through an administrative screening, an application validation and a review. At each stage, the submission is checked to see if it meets Health Canada’s standards of acceptability and regulatory safety and effectiveness requirements. A medical device submission can be deemed unacceptable at any point in the review process. If this occurs, the sponsor will have to provide additional information or a clarification before the submission can advance. Otherwise, a refusal letter may be issued by Health Canada.3
When a submission reaches the end of the review process, a decision whether or not to provide regulatory approval will be made.
US: Registration process for medical devices
The type of review required by the FDA depends on the medical device classification. Devices that are exempt from the premarket notification or authorization requirements are subject to general controls, including the establishment registration and device listing and labelling, record-keeping and reporting requirements. For medical devices that are subject to the 510(k) notification process, the target review timeline is 90 days. For PMA applications for class III devices, the target review timeline is 180 days.4
De novo process
For lower-risk medical devices that do not have a predicate device (which results in an automatic class III designation), a sponsor may request a de novo classification of the product in order to reclassify it as class I or II. The request must be in writing and sent within 30 days from the receipt of the Non-Substantial Equivalence (NSE) determination. The request should include all of the following:
- A description of the medical device
- Labelling information
- Reasons for the recommended classification (as class I or II)
- Information to support the recommendation
The de novo process has a 150-day review period. If the FDA classifies the medical device as class I or II, the sponsor will then receive regulatory approval to market the medical device. This device can then be used as a predicate device for other firms to submit a 510(k).5
Third-party review
For certain medical devices, a sponsor may choose an FDA-accredited third party (an “Accredited Person”) to conduct the primary review of 510(k). After the primary review of the 510(k), the Accredited Person forwards their review, recommendation and the 510(k) to the FDA. The FDA then issues a final determination within 30 days.6
EU: Legally placing medical devices on the market
Before a medical device can be legally placed on the market in Europe, it must go through an appropriate “conformity assessment process” to establish that it meets all the essential requirements of the applicable EU Directives. This process ensures that the device as it is designed and subsequently manufactured will meet the essential requirements. This enables the manufacturer to make a formal declaration of conformity and apply the CE mark of conformityi to the device.
For low-risk medical devices (class I non-sterile, non-measuring), such as a tongue depressor or a colostomy bag, the manufacturer can make a declaration of conformity with the essential requirements based solely on a self-assessment, without needing the involvement of a Notified Body.ii
For medium- to high-risk medical devices (Classes IIa, IIb, III and some of Class I), the manufacturer must call on a Notified Bodyii to assess the conformity. The Notified Body may perform one or more tasks as listed in Table 2. To some degree, the manufacturer may choose their method for the conformity assessment of the device and/or manufacturing system, depending on the type of device and the level of associated risk. The end result is a certificate of conformity that enables the manufacturer to apply CE marking to the product.7
Table 2: Certification activities of Notified Bodies
| Quality system certification | Device certification |
| ● Full QA system certification:
Certification based on auditing against a quality system standard for design, production and final inspection (for example, ISO 13485) ● Production QA system certification: Certification based on auditing against a quality system standard for production and final inspection (for example, ISO 13485―design control) ● Product QA system certification: Certification based on auditing against a quality system standard for final inspection (for example, EN 46003) |
● EC Design-examination certification:
Certification based on the examination of the design dossier versus standards/essential requirements (paperwork review) ● EC type-examination certification: Certification of the design based on testing of physical samples versus standards/essential requirements ● EC verification certification: Certification of individual production batches based on the testing of the whole batch or of batch samples versus standards/acceptance criteria |
Figure 1: Conformity assessment options available to manufacturers, based on device classifications per the Regulation (EU) 2017/7458
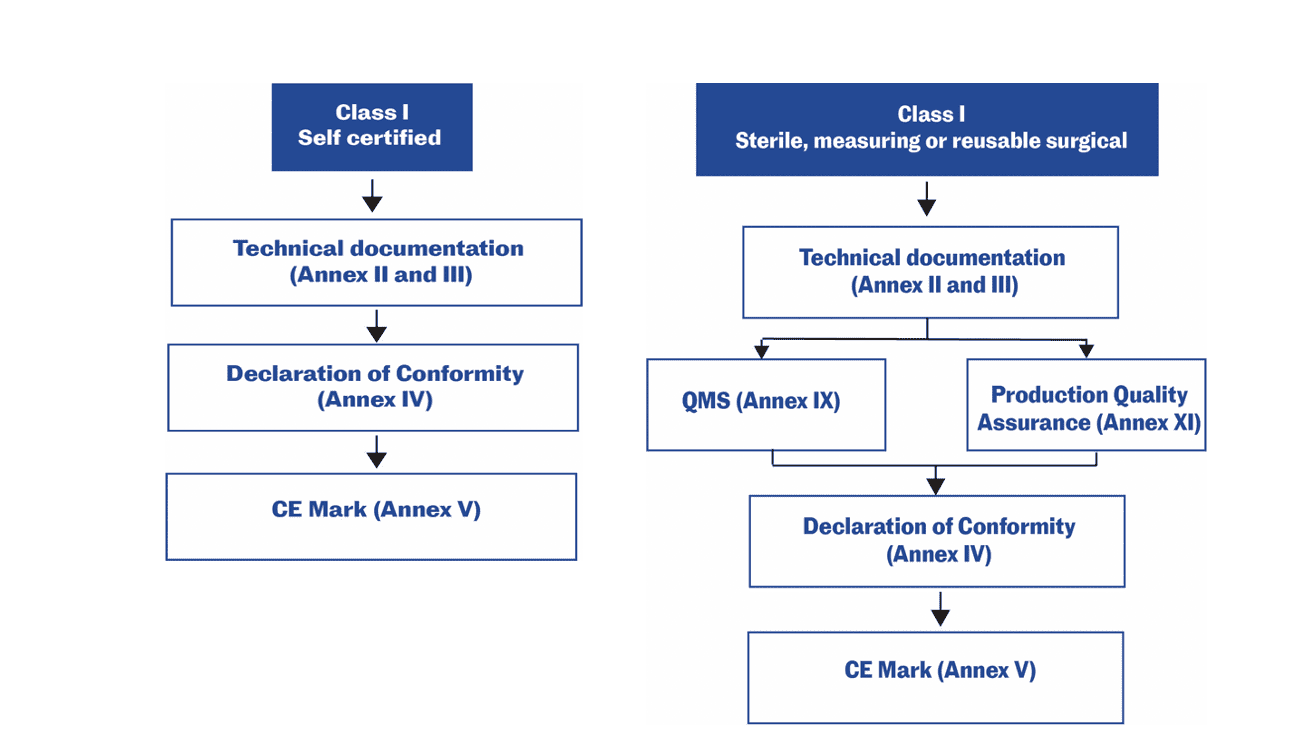
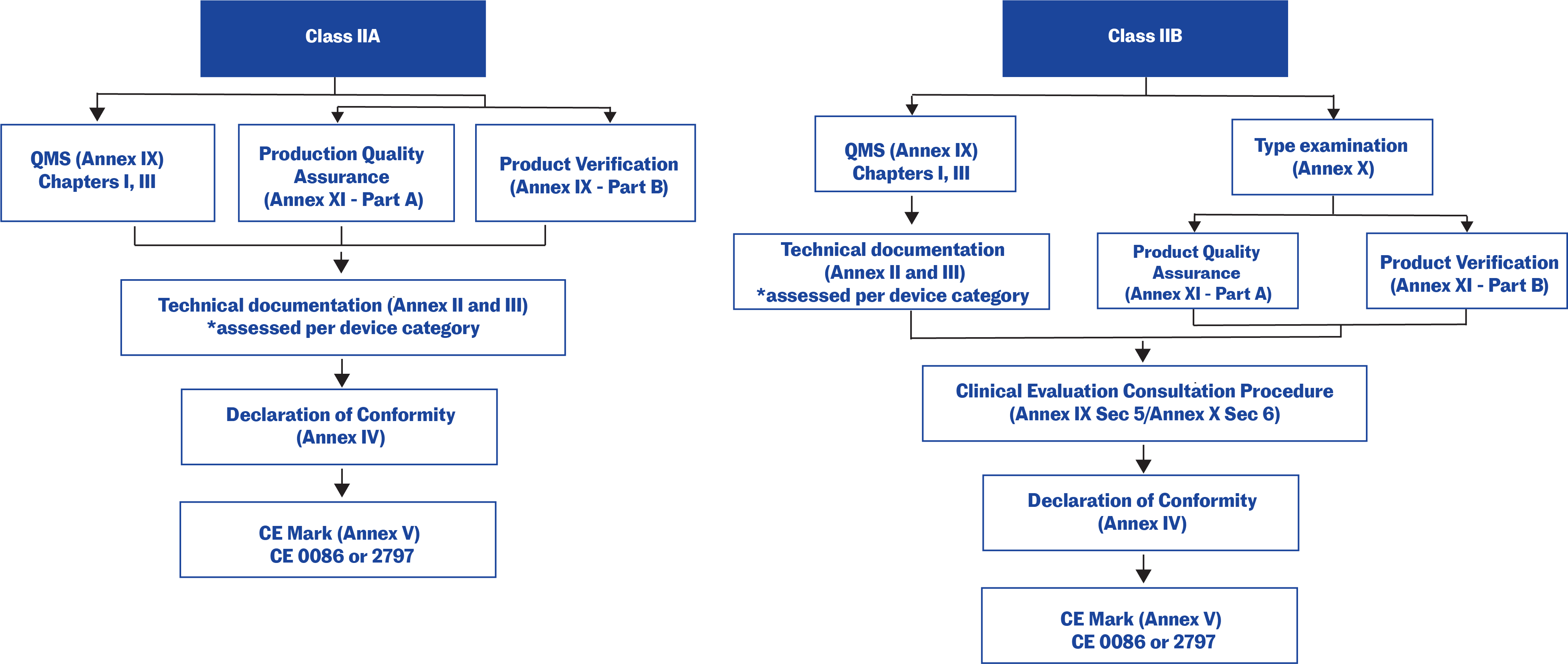
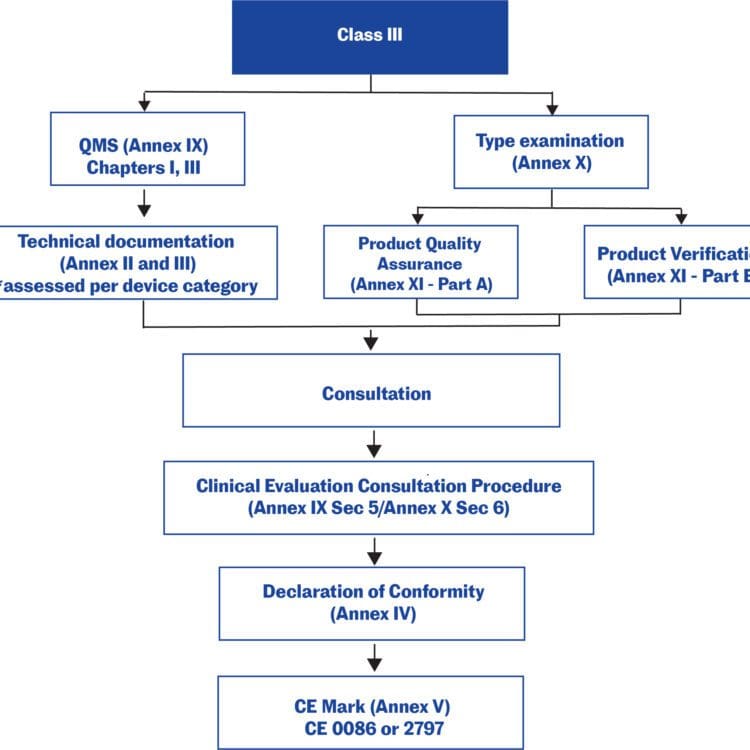
Figure 2: Transition timeline: MDD to MDR
![]()
Key changes in the new directive (MDD to MDR):
- Some devices are reclassified to higher-risk classes.
- Tighter areas of clinical investigations and post-market clinical follow-up.
- Notified Bodies have more responsibilities and some may lose their status as a Notified Body.
- Unique Device Identification (UDI) will be implemented.
- Several additions have been made to the EUDAMED database.9
The majority of Notified Bodies are independent commercial organizations that are designated, monitored and audited by the relevant Member States (of the EU) via their national Competent Authorities. A company is free to choose any Notified Body to cover the particular class of medical device under review. After approval, post-market surveillance is the responsibility of a Member State via the Competent Authority.
Manufacturers not located in the EU must appoint an authorized representative within the EU. After a product obtains the CE marking, its manufacturers must comply with any additional local registration requirements in each relevant Member State (such as informing the Competent Authority of their registered place of business or of any device description being placed on the market).
iA designation that indicates a product meets EU safety, health and environmental protection requirements.
iiA Notified Body is a certification organization that the national authority (the Competent Authority) of an EU Member State designates to carry out one or more of the conformity assessment procedures described in the annex(es) of the EU Directives. It must be qualified to perform all the functions set out in any annex for which it is designated.
Disclaimer
The information presented in these articles is intended to outline the general processes, principles and concepts of the healthcare product development lifecycle. Since regulatory requirements are ever-changing, it is current only as of the date of publication and not intended to provide detailed instructions for product development. Every healthcare product is unique and therefore so is its associated product development lifecycle. Specific advice should be sought from a qualified healthcare or other appropriate professional.
Updated: February 14, 2022
References
- Government of Canada. (2021, September 14). Frequently asked questions: Medical device establishment licensing and fees. Retrieved October 14, 2021, from https://www.canada.ca/en/health-canada/services/drugs-health-products/compliance-enforcement/establishment-licences/annual-review-documents/frequently-asked-questions-medical-device-establishment-licensing-fees.html#:~:text=A%20Medical%20Device%20Establishment%20Licence%20(MDEL)%20is%20required%20by%20Class,a%20medical%20device%20in%20Canada.
- U.S. Food and Drug Administration. (n.d.). Premarket approval (PMA). Retrieved October 14, 2021, from https://www.fda.gov/medical-devices/premarket-submissions/premarket-approval-pma.
- Petty, N.L. (2004). Chapter 6. Medical device submissions. In 2004 Fundamental of Canadian regulatory affairs. Washington: Regulatory Affairs Professional Society.
- Sucher, J.F., Jones, S.L. & Montoya, I.D. (2009). An overview of FDA regulatory requirements for new medical devices. Expert Opinion Med. Diagn. 3(1): 5-11.
- U.S. Food and Drug Administration. (n.d.). De Novo Classification Request. Retrieved November 4, 2021, from https://www.fda.gov/medical-devices/premarket-submissions/de-novo-classification-request.
- U.S. Food and Drug Administration. (n.d.). 510(K) third party review program. Retrieved November 4, 2021, from https://www.fda.gov/medical-devices/premarket-submissions/510k-third-party-review-program.
- Sorrel, S. (2006, August 1). Medical Device Development: U.S. and EU Differences. Less stringent requirements in the European Union result in faster medical device approval times. Applied Clinical Trial Online. Retrieved November 4, 2012, from http://www.appliedclinicaltrialsonline.com/appliedclinicaltrials/article/articleDetail.jsp?id=363640.
- BSI. (n.d.). Medical Device Regulation (MDR) Conformity Assessment Routes guide. Retrieved October 14, 2021, from https://www.bsigroup.com/en-GB/medical-devices/our-services/MDR-Revision/mdr-conformity-assessment-guide/.
- European Commission. (n.d.). EUDAMED – European Database on Medical Devices. Retrieved November 4, 2021, from https://ec.europa.eu/tools/eudamed/#/screen/home.